W ramach naszej witryny stosujemy pliki cookies w celu świadczenia Państwu usług na najwyższym poziomie, w tym w sposób dostosowany do indywidualnych potrzeb. Korzystanie z witryny bez zmiany ustawień dotyczących cookies oznacza, że pliki opisane w Polityce Prywatności będą zamieszczane na Państwa urządzeniu końcowym. W każdej chwili możecie Państwo zmienić ustawienia dotyczące plików cookies w przeglądarce internetowej. Akceptacja niezbędnych plików cookies jest wymagana do prawidłowego działania witryny. Szczegółowe informacje znajdą Państwo w zakładce POLITYKA PRYWATNOŚCI.
Jednym z głównych celów naszego laboratorium jest badanie ustanawiania latencji HIV w powiązaniu ze stochastyczną transkrypcją HIV oraz funkcjonalnym genomem gospodarza. Korzystając z ilościowych podejść genomicznych oraz metod opartych na uczeniu maszynowym, opracowanych w naszym laboratorium, dążymy do pogłębienia zrozumienia, w jaki sposób zmienność na poziomie pojedynczego wirusa w integralności genomu HIV i transkrypcji wpływa na konfigurację rezerwuarów HIV. Z tym celem powiązane są nasze kierunki badań:
1. Rola transkryptów antysensownych HIV (AST) w ustanawianiu latencji
Choć istnienie HIV AST i kodowanych przez nie białek zostało po raz pierwszy zasugerowane w analizach komputerowych w 1988 roku, podstawowe mechanizmy leżące u podstaw HIV AST pozostają słabo poznane. Badania in vitro wykazały, że antysensowne RNA HIV mogą sprzyjać inicjacji i utrzymywaniu latencji HIV; jednak dowody in vivo są ograniczone i sprzeczne, co pozostawia niepewność co do ich roli w patogenezie.
Naszym celem jest scharakteryzowanie potencjalnego progu stosunku między transkrypcją sensowną a antysensowną HIV poprzez określenie fenotypów transkrypcyjnych HIV. Dokonujemy tego, wykorzystując znakowany kodami kreskowymi HIV oraz klony komórkowe, z których każdy charakteryzuje się unikalnym fenotypem transkrypcyjnym HIV.
Publikacja powiązana: Więcek, K. i in. 2025. Computational and Structural Biotechnology Journal DOI:10.1016/j.csbj.2025.08.003
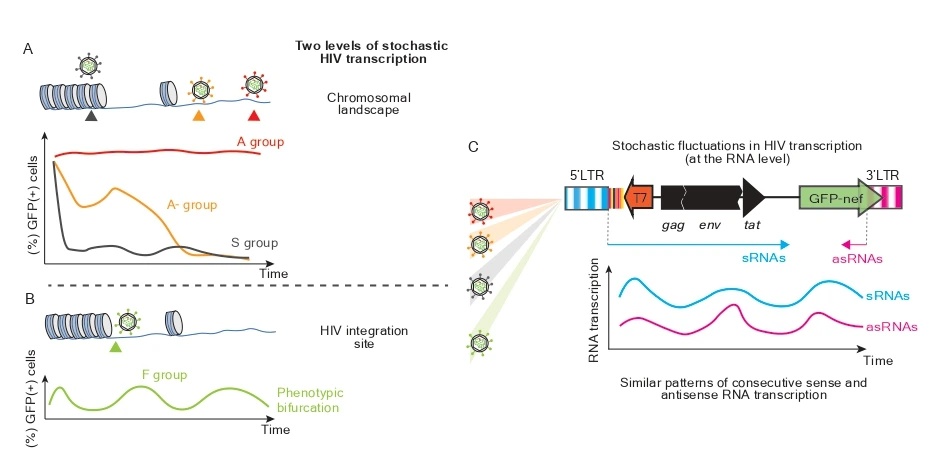
Rysunek 1. Udział asRNA HIV w stochastycznych fluktuacjach ekspresji genów HIV. Proponujemy, że fluktuacje transkrypcji HIV mogą występować na dwóch głównych poziomach: (1) krajobrazu chromosomalnego oraz (2) miejsca integracji HIV (A, B). Zaobserwowaliśmy minimalny stopień dziedziczenia takich fluktuacji i zidentyfikowaliśmy miejsca integracji prowirusów w pobliżu genomowych regionów powtórzonych, związanych z silnymi sygnałami H3K27ac i H3K4me3. W obu przypadkach zaobserwowano podobne czasowe wzorce ekspresji transkryptów sensownych i antysensownych (C), co sugeruje, że o stochastycznym fenotypie transkrypcji prowirusowej decyduje niezależna produkcja obu transkryptów, a nie ich proporcje.
2. Odrębna topologia wywołanych zadaniowo funkcjonalnych sieci genomowych w rezerwuarach HIV
Nasze podejście do rezerwuarów HIV zakłada, że mogą one być reprezentowane jako własności topologiczne (wywołane zadaniowo) sieci składającej się z różnych społeczności genów atakowanych przez HIV. Społeczności te tworzą tzw. sygnatury immunologiczne. Częstość integracji HIV w obrębie sieci może być wykorzystana jako wskaźnik do określania specyficznych typów komórek odpornościowych oraz prozapalnych czynników rozpuszczalnych, co ułatwia precyzyjne dostrajanie mikrośrodowiska rezerwuarów.
Aby pogłębić nasze zrozumienie heterogenicznych rezerwuarów HIV i ich implikacji funkcjonalnych, jako pierwsi zastosowaliśmy podejście konwergencyjne do charakteryzowania ich składu. W oparciu o narzędzia teorii grafów obserwujemy zauważalne cechy topologiczne w sieciach wzbogaconych w sygnatury immunologiczne, zawierające geny z prowirusami intaktnymi i uszkodzonymi, porównując osoby zakażone HIV-1 leczone terapią antyretrowirusową (ART) z elitarnymi kontrolerami.
Kluczowa zmienna – współczynnik wzbogacenia (rich factor) – odgrywa decydującą rolę w klasyfikacji odmiennych właściwości topologicznych sieci. Ekspresja genów gospodarza zwiększa dokładność klasyfikacji pomiędzy elitarnymi kontrolerami a pacjentami leczonymi ART. Modelowanie metodą łańcuchów Markowa Monte Carlo, zastosowane do symulacji różnych sieci grafowych, wykazało istnienie bariery wewnętrznej pomiędzy elitarnymi kontrolerami a osobami niebędącymi kontrolerami. Nasze badania stanowią ważny przykład wykorzystania podejść genomicznych wraz z narzędziami matematycznymi do zrozumienia złożoności rezerwuarów HIV.
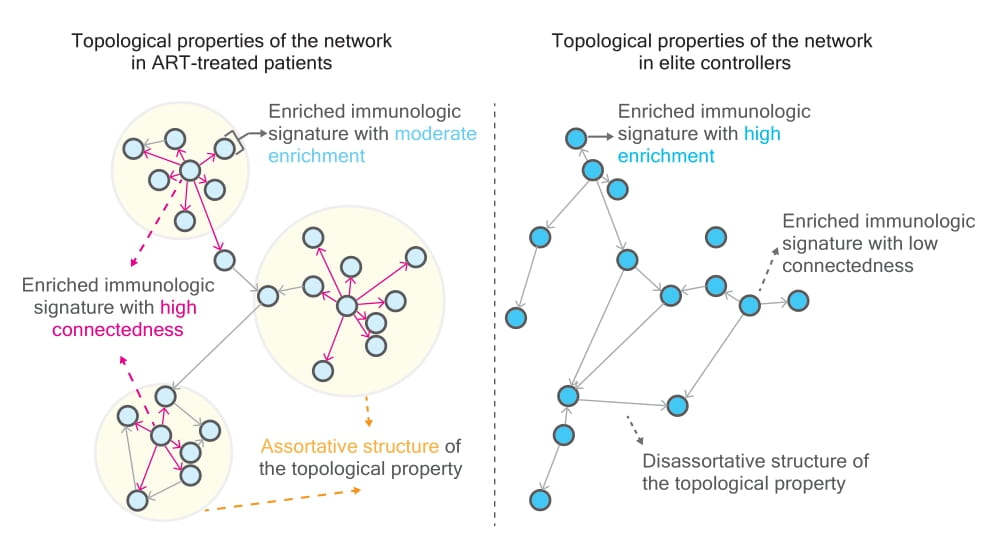
Rysunek 2. Topologia sieci rezerwuarów HIV u pacjentów leczonych ART w porównaniu z elitarnymi kontrolerami. W porównaniu z elitarnymi kontrolerami, architektura sieci u pacjentów leczonych ART wykazuje trzy główne cechy: (1) mniej intensywne wzbogacenie sygnatur, (2) wysoki stopień asortatywności oraz (3) zwiększone połączenia pomiędzy sąsiednimi wierzchołkami. Wyniki te sugerują, że architektura sieci u pacjentów leczonych ART jest bardziej spójna i strukturalna.
W naszym laboratorium badamy również infekcje wirusami odzwierzęcymi. Analizujemy związek pomiędzy wewnętrzną tropizacją wariantów koronawirusa a udomowieniem gospodarza.
3. Identyfikacja potencjalnych markerów genetycznych SARS-CoV-2 wynikających z udomowienia gospodarza
Opracowaliśmy pipeline oparty na k-merach, nazwany Pathogen Origin Recognition Tool using Enriched K-mers (PORT-EK), służący do identyfikacji regionów genomowych wzbogaconych u odpowiednich gospodarzy po porównaniu metagenomów izolatów między dwoma gatunkami gospodarzy.
Za jego pomocą zidentyfikowaliśmy tysiące k-merów wzbogaconych u północnoamerykańskich jeleni wirginijskich oraz betakoronawirusów w porównaniu z izolatami ludzkimi. Ponadto wykazaliśmy odmienne krajobrazy pokrycia k-merów wzbogaconych u jeleni i nietoperzy oraz odkryliśmy 144 mutacje w k-merach wzbogaconych, uzyskanych z porównania metagenomów wirusowych pomiędzy izolatami nietoperzy i ludzi.
Zaobserwowaliśmy również, że trzecia pozycja w kodonie genetycznym jest szczególnie podatna na mutacje, co prowadzi do wysokiej częstości mutacji synonimicznych aminokwasów zachowujących te same właściwości fizykochemiczne co aminokwasy niezmienione. Co istotne, jesteśmy w stanie klasyfikować i przewidywać prawdopodobieństwo przynależności do gatunku gospodarza na podstawie liczby wzbogaconych k-merów.
Publikacja powiązana: Wiśniewski and Chen. 2024. iMetaOmics DOI:10.1002/imo2.70019
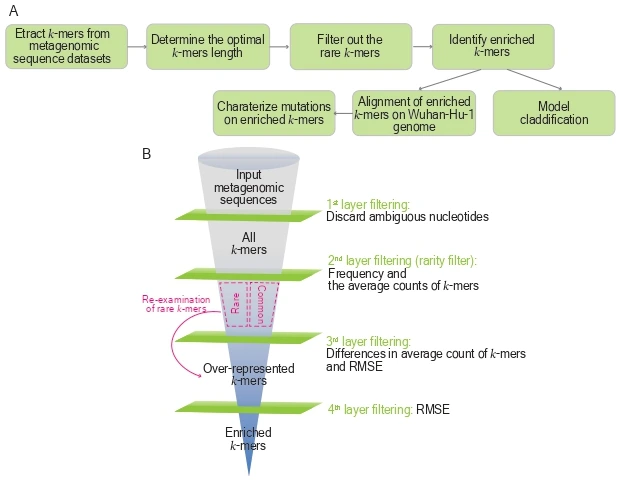
Rysunek 3. Racjonalny projekt PORT-EK i określenie wzbogaconych k-merów. (A) Schemat analityczny PORT-EK. PORT-EK składa się z czterech kroków: (1) przygotowania macierzy k-merów, (2) filtrowania i wyboru k-merów, (3) identyfikacji mutacji specyficznych dla gospodarza oraz (4) klasyfikacji gospodarzy. Szczegóły opisano w tekście głównym. (B) Wykres lejkowy przedstawiający strategie filtrowania do wyboru wzbogaconych k-merów. W pipeline PORT-EK stosuje się cztery poziomy filtrowania.