As part of our website, we use cookies to provide you with the highest level of service, including services tailored to your individual needs. By continuing to use the website without changing your cookie settings, you agree to the placement of cookies on your device as described in our Privacy Policy. You can change your cookie settings in your web browser at any time. Acceptance of essential cookies is required for the proper functioning of the website. For more detailed information, please refer to our PRIVACY POLICY.
One of the main goals of our laboratory is to study the establishment of HIV latency in connection with stochastic HIV transcription and the functional host genome. Using quantitative genomic approaches and machine learning–based methods developed in our lab, we aim to deepen our understanding of how single-virus-level variability in HIV genome integrity and transcription influences the configuration of HIV reservoirs. Our research directions include the following:
1. The Role of HIV Antisense Transcripts (ASTs) in Latency Establishment
Although the existence of HIV antisense transcripts (ASTs) and their encoded proteins was first suggested by computational analyses in 1988, the underlying mechanisms remain poorly understood. In vitro studies have shown that HIV antisense RNAs may promote the initiation and maintenance of HIV latency; however, in vivo evidence is limited and contradictory, leaving their role in pathogenesis uncertain.
Our goal is to characterize the potential threshold ratio between sense and antisense HIV transcription by defining distinct HIV transcriptional phenotypes. We achieve this using barcode-labeled HIV constructs and cell clones, each exhibiting a unique HIV transcriptional profile.
Related publication: Więcek, K. i in. 2025. Computational and Structural Biotechnology Journal DOI:10.1016/j.csbj.2025.08.003
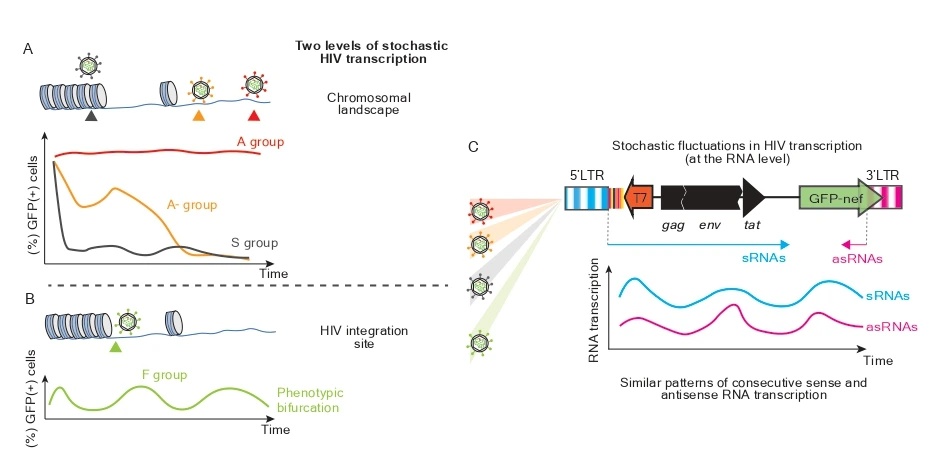
Figure 1. The contribution of HIV asRNA to stochastic fluctuations in HIV gene expression. We propose that stochastic HIV transcription may occur at two main levels: (1) the chromosomal landscape and (2) the HIV integration site (A, B). We observed a minimal degree of heritability of such fluctuations and identified proviral integration sites near genomic repeat regions associated with strong H3K27ac and H3K4me3 signals. In both cases, similar temporal patterns of sense and antisense transcript expression were observed (C), suggesting that stochastic HIV transcription phenotypes are determined by independent production of both transcript types rather than their ratio.
2. Distinct Task-Induced Topology of Functional Genomic Networks in HIV Reservoirs
Our approach to HIV reservoirs assumes that they can be represented as task-induced topological properties of networks composed of gene communities targeted by HIV. These communities form immunological signatures. The frequency of HIV integration within these networks can serve as an indicator for identifying specific immune cell types and soluble proinflammatory factors, facilitating precise tuning of the reservoir microenvironment.
To better understand the heterogeneity and functional implications of HIV reservoirs, we were among the first to apply a convergent approach to characterize their composition. Using graph theory–based tools, we observed distinct topological features in networks enriched for immunological signatures containing genes with intact and defective proviruses, comparing ART-treated individuals with elite controllers.
A key variable—the rich factor—plays a decisive role in classifying distinct topological network properties. Host gene expression improves classification accuracy between elite controllers and ART-treated patients. Using Markov Chain Monte Carlo modeling, we simulated various graph networks and discovered an internal barrier separating elite controllers from non-controllers. Our research represents an important example of using genomic and mathematical approaches to unravel the complexity of HIV reservoirs.
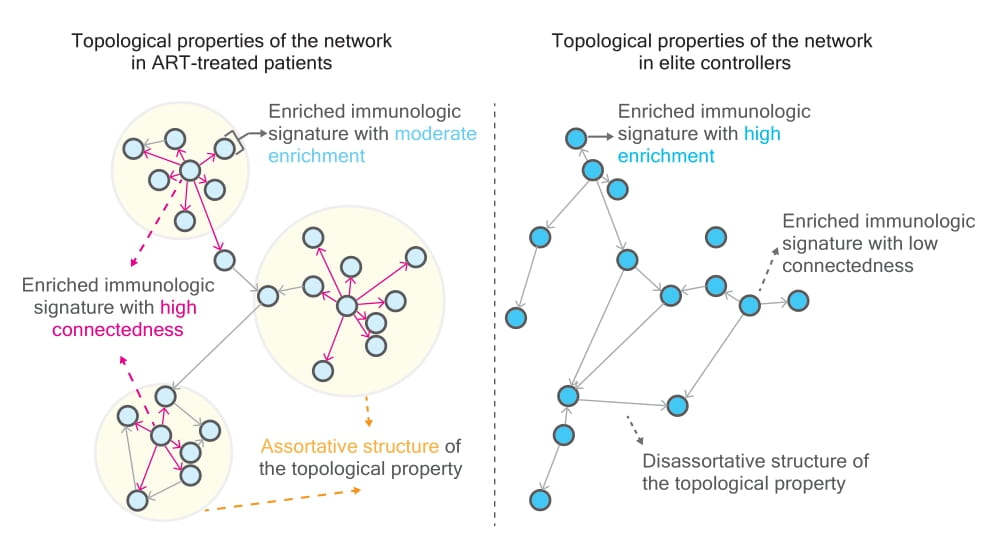
Figure 2. Topology of HIV reservoir networks in ART-treated patients versus elite controllers. Compared with elite controllers, the network architecture of ART-treated patients shows three major features: (1) weaker signature enrichment, (2) higher assortativity, and (3) increased connectivity between adjacent nodes. These results suggest that the networks in ART-treated patients are more cohesive and structured.
We also study zoonotic viral infections, analyzing the relationship between intra-host tropism of coronavirus variants and host domestication.
3. Identification of Potential Genetic Markers of SARS-CoV-2 Resulting from Host Domestication
We developed a k-mer–based pipeline, called PORT-EK (Pathogen Origin Recognition Tool using Enriched K-mers), designed to identify genomic regions enriched in specific hosts by comparing metagenomes of isolates between two host species.Opracowaliśmy pipeline oparty na k-merach, nazwany Pathogen Origin Recognition Tool using Enriched K-mers (PORT-EK), służący do identyfikacji regionów genomowych wzbogaconych u odpowiednich gospodarzy po porównaniu metagenomów izolatów między dwoma gatunkami gospodarzy.
Using PORT-EK, we identified thousands of enriched k-mers in North American white-tailed deer and betacoronaviruses compared to human isolates. We further demonstrated distinct coverage landscapes of enriched k-mers in deer and bats and discovered 144 enriched k-mer mutations derived from comparisons between bat and human viral metagenomes.
We also observed that the third codon position is particularly susceptible to mutations, leading to a high frequency of synonymous amino acid substitutions that preserve the same physicochemical properties as the original residues. Importantly, we can classify and predict host species identity based on the number of enriched k-mers.
Related publication: Wiśniewski and Chen. 2024. iMetaOmics DOI:10.1002/imo2.70019
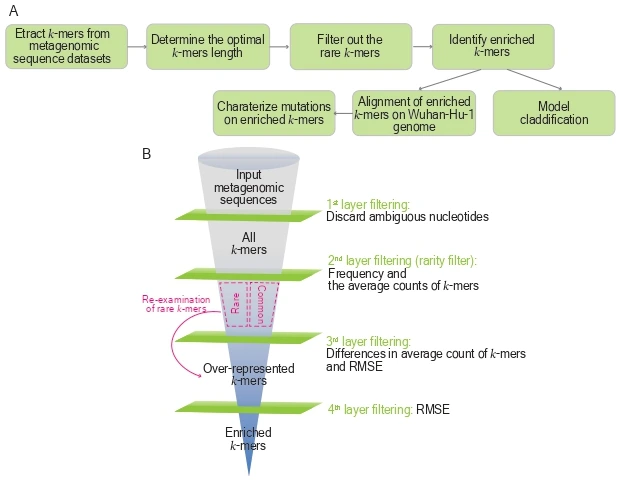
Figure 3. Schematic design of PORT-EK and identification of enriched k-mers. (A) Analytical workflow of PORT-EK, which consists of four steps: (1) k-mer matrix preparation, (2) k-mer filtering and selection, (3) identification of host-specific mutations, and (4) host classification. (B) Funnel chart illustrating the filtering strategies used to select enriched k-mers. Details are described in the main text.