Quantitative Virology Research Group

Our research is bound to molecular barcodes, which are molecular tags, enabling the achievement of studies with resolution of single viruses. Together with in silico modeling and AI-based approaches, we investigate the mechanistic interplay between viruses, i.e. HIV-1 and SARS-CoV-2, and the host genome, with an emphasis on:
- the investigation of latent HIV-1 reservoirs and stochastic HIV-1 transcription and
- the identification of functional annotations of SARS-CoV-2 mutational signatures responsible for cross-species transmission.
- Lab Twitter: @BhiveLaboratory
- Lab webpage: https://hengchangchen.wixsite.com/silence/home
Funders:
![]()
Heng-Chang Chen, Dr. rer. nat., principal investigator
I am a molecular virologist and data scientist with over eight years of experience. I obtained a Ph.D. degree in Microbiology from the Humboldt University of Berlin (Berlin, Germany). Afterward, I joined the laboratory of Dr. Filion at the Centre for Genomic Regulation (Barcelona, Spain) and the laboratory of Dr. Benkirane at the Institute of Human Genetics (Montpellier, France) for postdoc research. During this period, I commenced my studies in HIV. In November 2022, I was appointed as the Junior Research Group Leader to head the new virology laboratory in the Center for Population Diagnostics in the Łukasiewicz Research Network – PORT Polish Center for Technology Development (Wrocław, Poland).

Janusz Wiśniewski. Ph.D. Postdoc
I am a Bioinformatician with a biochemical past. I spent most of my career studying protein structure and interactions, both in academia and industry. My current research focuses on using language models with genomic and protein sequences of viruses, but my interests cover all applications of machine learning in life sciences.

Kamil Więcek. M.Sc. Process Engineer
I am a graduate of the University of Wrocław where I majored in medical biotechnology. Most of my scientific work so far has been focused on Immunology and inflammatory diseases. At present, I attempt to dissect the mechanistic interplay between the 3D genome organization and insert-specific HIV transcription.

Oliwia Filas, M.Sc. Process Engineer
I am biotechnologist and most of my work so far has been focused both on molecular biology and microbiology. Now I take part in the project that studies how the HIV antisense transcripts regulate HIV transcription.
Lab alumni
Jędrzej Mazur, Intern, 03/2023-06/2023. Currently: Ph.D. student, Center for Structural Systems Biology, Leibniz Institute of Virology, Germany.
One of the main objectives of our laboratory is to study the establishment of HIV latency coupled with stochastic HIV transcription and the host functional genome.
Using quantitative gnomic and machine-learning-based approaches tailored from the laboratory, we aim to deepen our grasp on how variations, with resolution of single viruses, in HIV genome integrity and transcription influence the configuration of HIV reservoirs. Two research lines aligning with this objective are conducted.
- Role of HIV antisense transcripts (AST) in the establishment of latency
Although the existence of HIV AST and encoded proteins was first postulated by computer analyses in 1988, at present the fundamental mechanisms behind HIV AST are not fully understood. HIV antisense RNAs have been demonstrated in vitro to be capable of promoting the initiation and maintenance of HIV latency; however, HIV AST in vivo is poorly detected and the observations are contradictory, leaving uncertainty to conclude the mechanism of how HIV AST contributes to its pathogenesis. We characterize the possible threshold of the ratio between HIV sense and antisense transcription, determining the transcriptional phenotypes of HIV based on barcoded HIV and cellular clones possessing unique transcriptional phenotypes of HIV.
- Distinguishable topology of the task-evoked functional genome networks in HIV reservoirs
Our len to view HIV reservoir is that it can be represented as the topological (task-evoked) property of a network consisting of different communities of the genes targeted by HIV. Such communities are so-called immunologic signatures. HIV integration frequency within a network might be used as a proxy to define specific immune cell types and proinflammatory soluble factors, facilitating fine-tuning of the microenvironment of reservoirs. To deepen our understanding of such heterogeneous HIV reservoirs and their functional implications, we pioneer the integration of a convergence approach to characterize the composition of HIV reservoirs.
Based on graph-theoretical tools, we observe noticeable topological properties in networks, featuring immunologic signatures enriched by genes harboring intact and defective proviruses, when comparing antiretroviral therapy (ART)-treated HIV-1-infected individuals and elite controllers. The key variable, the rich factor, plays a pivotal role in classifying distinct topological properties in networks. The host gene expression strengthens the accuracy of classification between elite controllers and ART-treated patients. Markov chain Monte Carlo modeling for the simulation of different graph networks demonstrated the presence of an intrinsic barrier between elite controllers and non-elite controllers. Notably, our work provides a prime example of leveraging genomic approaches alongside mathematical tools to unravel the complexities of HIV reservoirs.
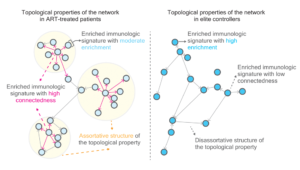
Figure 1. The network topology of HIV reservoirs in ART-treated patients versus elite controllers. In comparison to elite controllers, the network architecture in ART-treated patients exhibits three key characteristics (1) a less intense magnitude of signature enrichment, (2) a high degree of assortativity, and (3) elevated connectedness between two adjacent vertices. These findings suggest that the network architecture is more connective and structural in ART-treated patients.
Publications related to this research line:
- Chen, H.-C. 2023. Vaccines DOI:10.3390/vaccines11020402
- Więcek, K., and Chen, H.-C. 2023. iScience DOI:10.1016/j.isci.2023.108342
- Wiśniewski, et al. 2024. BioRxiv DOI:10.1101/2024.02.05.578936
Another scientific objective in the laboratory is to study the linkage between the intrinsic tropisms of coronavirus variants and host domestication.
- Identification of potential SARS-CoV-2 genetic markers resulting from host domestication
We develop a k-mer-based pipeline, namely the Pathogen Origin Recognition Tool using Enriched K-mers (PORT-EK) to identify genomic regions enriched in the respective hosts after the comparison of metagenomes of isolates between two host species. Using it we successfully identify thousands of k-mers enriched in US white-tailed deer and betacoronaviruses while comparing them with human isolates. In addition, we demonstrate different coverage landscapes of k-mers enriched in deer and bats and unravel 144 mutations in enriched k-mers yielded from the comparison of viral metagenomes between bat and human isolates. Additionally, we observe that the third position within a genetic codon is prone to mutations, resulting in a high frequency of synonymous mutations of amino acids harboring the same physicochemical properties as unaltered amino acids. Importantly, we are able to classify and predict the likelihood of host species based on the enriched k-mer counts.
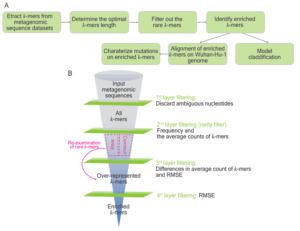
Figure 2. Rational design of PORT-EK and determination of the enriched k-mers. (A) The analytical pipeline of PORT-EK. PORT-EK consists of four steps including (1) k-mers matrix preparation, (2) k-mers filtering and selection, (3) the identification of host-specific mutations, and (4) the classification of hosts. Details are described in the main text. (B) Funnel plot representing the filtering strategies for the selection of enriched k-mers. Four layers of filtering are applied in the PORT-EK pipeline.
Publications related to this research line:
1. Wiśniewski and Chen. 2024. BioRxiv DOI:10.1101/2024.07.27.605454
Our funders
![]()
SONATA BIS 12, 01/10/2023-30/09/2027
International Multilateral Partnerships for Resilient Education and Science System in Ukraine (IMPRESS-U), 2024-2025
- Tsalsabila, A., V.A. Dabur, I.J. Budiarso, S. Wustoni, H.-C. Chen, M.D. Birowosuto, A. Wibowo, S. Zeng. 2024.
Progress and Outlooks in Designing Photonic Biosensor for Virus Detection.
Adv Opt Mater. DOI:10.1002/adom.202400849
2023
- Thenin-Houssier, S., Machida, S., Jahan, C., Bonnet-Madin, J., Abbou, S., Chen, H.-C., Tesfaye, R., Cuvier, O., and Benkirane, M. 2023.
POLE3 is a transcriptional repressor of unintegrated linear HIV-1 DNA required for efficient virus integration and escape from innate immune sensing.
Sci. Adv. DOI:10.1126/sciadv.adh3642 - Więcek, K., and Chen, H.-C. 2023.
Understanding latent HIV-1 reservoirs through host genomics approaches.
iScience DOI:10.1016/j.isci.2023.108342 - Chen, H.-C. 2023.
A systematic review of the barcoding strategy that contributes to COVID-19 diagnostics at a population level.
Front. Mol. Biosci. DOI:10.3389/fmolb.2023.1141534 - Chen, H.-C. 2023.
The Dynamic Linkage between Provirus Integration Sites and the Host Functional Genome Property Alongside HIV-1 Infections Associated with Antiretroviral Therapy.
Vaccines DOI:10.3390/vaccines11020402
2022
- Chen, H.-C. 2022.
Position matters – a discussion of HIV gene expression (中文主題名稱:原來擺對位置很重要 — 淺談人類免疫缺陷病毒的基因表現).
Science Education Monthly (科學教育月刊) ISSN 1021-3708, 454, P25-32, DOI:10.6216/SEM
2021
- Lucic, B., H.-C. Chen, M. Kuzman*, E. Zorita*, J. Wegner, V. Minneker, W. Wang, R. Fronza, M. Schmidt, and R. Stadhouders, V. Roukos, K. Vlahovicek, G. Filion and M. Lusic. 2021.
Author Correction: Spatially clustered loci with multiple enhancers are frequent targets of HIV-1.
Nat. Commun. doi.org/10.1038/s41467-021-26471-w
2020
- Vansant, G., H.-C. Chen, E. Zorita, Trejbalová, K., Miklík, D., G. Filion, and Z. Debyser. 2020.
The chromatin landscape at the HIV-1 provirus integration site determines viral expression.
Nucleic Acids Res. doi.org/10.1093/nar/gkaa536 - Machida, S., D. Depierre, H.-C. Chen, S. Houssier, G. Petitjean, C. Doyen, M. Takaku, O. Cuvier and M. Benkirane. 2020.
Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome.
PNAS doi: 10.1073/pnas.1913754117
2019
- Lucic, B.*, H.-C. Chen*, M. Kuzman*, E. Zorita*, J. Wegner, V. Minneker, W. Wang, R. Fronza, M. Schmidt, and R. Stadhouders, V. Roukos, K. Vlahovicek, G. Filion and M. Lusic. 2019.
Spatially clustered loci with multiple enhancers are frequent targets of HIV-1.
Nat. Commun. doi: 10.1038/s41467-019-12046-3
2018
- Abner, E., M. Stoszko, L. Zeng, H.-C. Chen, A. Izquierdo-Bouldstridge, T. Konuma, E. Zorita, E. Fanunza, Q. Zhang, T. Mahmoudi, M.-M. Zhou, G. Filion, and A. Jordan. 2018.
A new quinoline BRD4 inhibitor targets a distinct latent HIV-1 reservoir for re-activation from other ‘shock’ drugs.
J Virol. doi: 10.1128/JVI.02056-17. - Chen, H.-C.*, E. Zorita, and G. Filion*. 2018.
Using Barcoded HIV Ensembles (B-HIVE) for single provirus transcriptomics.
Curr Protoc Mol Biol. doi: 10.1002/cpmb.56
2017
- Chen, H.-C., J.P. Martinez, E. Zorita, A. Meyerhans, and G. Filion. 2017.
Position effects influence HIV latency reversal.
Nat Struct Mol Biol. doi: 10.1038/nsmb.3328.
2015
- Corrales, M., P. Cusco, D.R. Usmanova, H.-C. Chen, N.S. Bogatyreva, G.J. Filion, and D.N. Ivankov. 2015. Machine learning: how much does it tell about protein folding rates?
PLoS One doi:10:e0143166.
2008
- Chen, P.M., H.-C. Chen, C.T. Ho, C.J. Jung, H.T. Lien, J.Y. Chen, and J.S. Chia. 2008.
The two-component system ScnRK of Streptococcus mutans affects hydrogen peroxide resistance and murine macrophage killing.
Microbes and Infection doi:10:293-301.
We always welcome talented and highly motivated people (postdoc, PhD, and intern) who are enthusiastic about science and are interested in our research area. Please do not hesitate to contact us at heng-chang.chen@port.lukasiewicz.gov.pl.